
 Sophos Naked Security
Sophos Naked Security Securelist
Securelist cyber-secret futurist
cyber-secret futurist I'm, the bookworm
I'm, the bookworm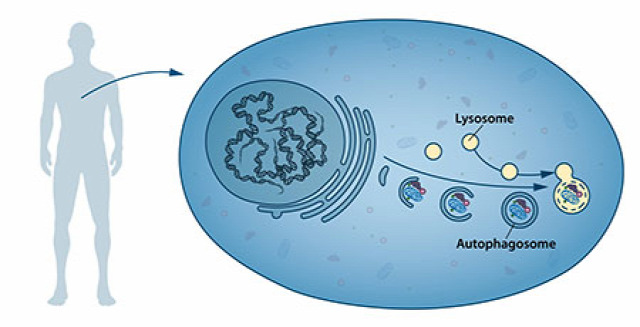 Ahogy arról hétfőn beszámoltam, idén az orvosi-fiziológiai Nobel-díjat az autofágia genetikai szabályozásának feltárásáért ítélték oda.
Ahogy arról hétfőn beszámoltam, idén az orvosi-fiziológiai Nobel-díjat az autofágia genetikai szabályozásának feltárásáért ítélték oda.
Az élő sejtekben, ugyan sejttípustól eltérő mértékben, de folyamatosan sérülnek, kiöregszenek, így egyszerűen javításra vagy ha az már nem megy, lebontásra szorulnak bizonyos kisebb struktúrák, az ún. organellumok avagy sejtszervecskék és az azokat alkotó makromolekulák. Abban az esetben, ha ez nem lenne garantált és óramű pontossággal szabályozott, felhalmozódna számos működésképtelenné vált organellum vagy éppen hibásan tekeredett fehérje, ez pedig nagyon gyorsan a sejt halálához vezetne.
A belső törmelékek lebontását külön, erre specializálódott organellumok végzik, amiket lizoszómáknak nevez a szakirodalom. A lizoszómák a fölöslegessé vált makromolekulákat egyre kisebb részeire, akár monomerjeire is bonthatják, közvetve segítik más organellumok újraépítését, esetleg egyszerűen eltüntetik, amit megemésztettek, de olyan is előfordulhat, hogy a sejt belsejének többi részétől elkülönített, memránnal határolt csomagban zárványként maradnak jelen. Olyan is előfordulhat, amikor a lizoszómába egy teljes organellum kerül bele, ezeket a struktúrákat nevezi a szakirodalom autofagoszómának.
Ahogy korábban is utaltam rá, sebészi pontossággal szükséges felismernie a sejtnek, hogy mik azok a részek, amiket le kell bontani, mik azok, amiket nem, majd ugyanilyen pontossággal levezényelni a teljes folyamatot, minderre pedig természetesen számos forgatókönyvet készített az evolúció.
A japán kutató penészgombákon keresztül azt tanulmányozta, hogy bizonyos gének kiütésével hogyan módosul az autofágia folyamata, ilyen módon szisztematikusan sikerült feltérképezni a szabályozásban szerepet játszó gének hálózatát. A gombák esetében talált gének analógjai vagy maguk a gének pedig megtalálhatók a növényvilág képviselői és az állatvilág legkülönbözőbb tagjai közt is, így az emberben szintén. Mindennek az orvosi jelentősség abban áll, hogy ismeretesek olyan betegségek, amikkel együtt jár az autofágia hibás működése, így a jövőben a közvetlen molekuláris medicina számára is fontos targetté válhat a félresiklott folyamatok helyreállítása.
Vannak esetek, amikor veleszületett rendellenességként a lizoszómák nem tudják lebontani vagy tárolni a szükséges anyagokat, amik még abban az esetben is súlyos tüneteket okozhatnak, ha egyébként csak bizonyos sejttípusok esetén jelentkeznek.
A Gaucher-kór esetén például csak a fehérvérsejtek egy bizonyos típusa, a makrofágok érintettek, amik nem tudnak lebontani egy bizonyos típusú makromolekulát. A kór egyik típusa nem okoz különösebben súlyos tüneteket és enzimterápiával kezelhető is, míg a másik két megjelenési formája rendszerint korai halált eredményez.
A Hunter-szindróma ugyancsak egyetlen enzim defektusára vezethető vissza, ami miatt a lizoszóma képtelen tárolni egy létfontosságú enzimet, ami miatt a dermatán szulfát és a heparán szulfát, létfontosságú struktúrfehérjék forgalma gátolt.
A szintén lizoszomális enzim hiánya okozta Fabry-kór esetén ugyancsak a teljes testet és persze életminőséget befolyásoló tünetek jelentkeznek, ebben az esetben emlékeim szerint az egyik galaktozidáz enzim teljes vagy részleges hiánya okozza a súlyos megbetegedést.
Eméleim szerint még legalább 20-30 betegség ismert, amikor a lizoszóma és ezzel együtt az autofágia valamilyen defektusa felelős súlyos tünetekért, az esetek többségében a lebontásra szánt makromoelkulák bejutnak ugyan a lizoszómába, a lebontó enzim hiánya miatt elbomlani már nem tudnak.
Kép: nobelprize.org